Machine learning
Developing machine learning and statistical techniques to model and optimize black-box systems and simulated processes.
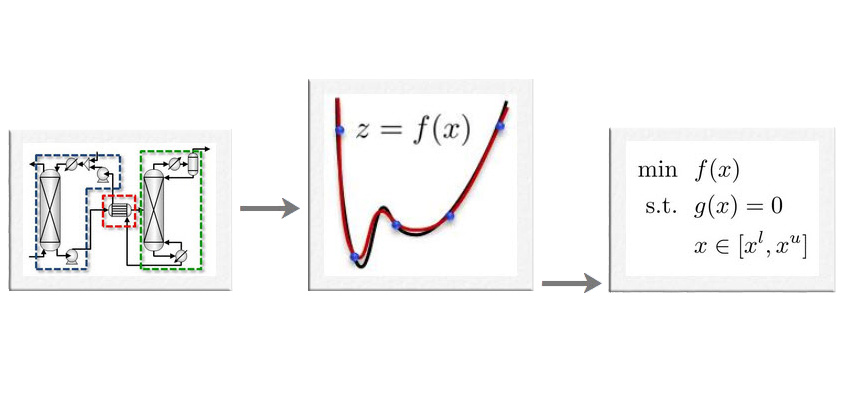
Developing machine learning and statistical techniques to model and optimize black-box systems and simulated processes.